Human Genome Project
Zusammenfassung
Im Februar des Jahres 2001 veröffentlichen zwei Gruppen von Wissenschaftlern Rohversionen der menschlichen DNA-Sequenz. Eine Gruppe war die private Firma „Celera Genomics", die damals noch unter der Leitung von Craig Venter stand, und die andere war das von öffentlicher Hand finanzierte „International Human Genome Sequencing Consortium". Im April 2003 konnten beide Gruppen nun das Endergebnis ihrer weiteren Bemühungen um Vervollständigung der Sequenz präsentieren: 98% der Sequenz des menschlichen Genoms sind bekannt. Beide benötigten mehrerer Milliarden Dollar für dieses Projekt. Aber welche Bedeutung haben nun dieses Kenntnisse? Wie viel wissen wir eigentlich und welche Gefahren können durch Missbrauch dieses Wissens entstehen?
Schlüsselwörter: Sequenzierung des menschlichen Genoms, Funktion
Abstract
In February 2001 two scientific groups published the draft sequence of the human genome. One group was the private company "Celera Genomics", which was lead by Craig Venter, and the other the public financed "International Human Genome Sequencing Consortium". In April 2003 the final result of further work of these two groups could be presented: 98% of the human Genome Sequence is now known. Both groups needed billions of dollars. But what could be done with this knowledge? What do we know actually and what dangers can come from misuse of this knowledge?
Keywords: Sequencing of the Human Genome, Function
Einleitung
Seitdem im Februar 2001 bekannt wurde, dass das menschliche Genom durchsequenziert worden ist, reißen die Diskussionen über den Wert und die Bedeutung dieser Kenntnis nicht ab. Dieses Jahr wurde die Diskussion neu angefacht durch die Nachricht, dass nun die gesamte Sequenz, oder zumindestens 98% bekannt ist.1 Schlagzeilen wie: „Die Natur des Menschen ist entschlüsselt" oder „Heilung aller Erbkrankheiten steht bevor", versetzen die einen in Freude und wecken bei den anderen Misstrauen.
Seit Beginn des Sequenzierprojekts gab es Wissenschaftler, die behaupteten, dass dieses Vorhaben nur Verschwendung von Ressourcen ist, dass die gewonnene Information zu gering sei, um sich dermaßen in Unkosten zu stürzen und das Talent junger Forscher zu verschwenden. Viele ihrer Kollegen geben ihnen heute insofern Recht, als sie meinen, dass die Sequenzierung erst der erste Schritt in die Richtung des Verständnisses der genetischen Zusammenhänge im Menschen sei.
In diesem Artikel soll zunächst eine kleine Einführung über die Geschichte des Genomprojekts und die technischen Hintergründe des „modernen" Sequenzierens gegeben werden. (Einen Überblick über die technischen Möglichkeiten, die in der Anfangszeit des humanen Genomprojekts zur Verfügung standen, gibt die Imabe-Dokumentation: „Humanes Genomprojekt" aus dem Jahr 1991.2) Danach soll auf Aussagen, die nun mit der humanen Sequenz gemacht werden können, und auf weitere Forschungen, die sich aus dem Sequenzierprojekt ergeben, eingegangen werden.
Die Sequenzierung des menschlichen Genoms
Bereits in der Mitte der 80er Jahre des 20. Jahrhunderts gab es drei verschiedene Wissenschaftler bzw. wissenschaftliche Arbeitsgruppen, die die Sequenzierung des menschlichen Genoms als sehr wichtig einstuften. Die erste dieser Gruppen war das US Department of Energy (DOE), das 1984 ein Treffen in Alta veranstaltete. Dort ging es um die Auffindung von Möglichkeiten, mit denen vererbte Schädigungen (durch Strahlung oder ähnliches) nachgewiesen werden können. Die einzige Methode schien die Durchsequenzierung des menschlichen Genoms zu sein, um so die Veränderung in den wenigen betroffenen Basen entdecken zu können.3
1985 organisierte der damalige Kanzler der Universität von Kalifornien in Santa Cruz, Robert Sinsheimer, ein Treffen einiger Gruppen an seiner Universität. Ziel war es, auf diese Weise einen Großteil des Projekts in der Nähe der Universität durchführen zu können, indem die teilnehmenden Gruppen zur „Colaboration" eingeladen wurden.4 Der dritte dieser „Pioniere" des Genomprojekts war Robert Dulbecco, der 1986 einen Artikel verfasste, in dem er die Sequenzierung des menschlichen Genoms für unumgänglich für die Weiterentwicklung der Krebsforschung hielt.5
1988 konnte dann mit der Realisierung dieses Großprojekts begonnen werden. Das Symposium „Molecular Biology of homo sapiens" im Jahre 1988 gab den letzten Anstoß dazu. Das NIH (National Institute for Health) stieg ebenfalls zu dieser Zeit ein. Ein eigenes Büro für die „Human Genome Research" wurde eingerichtet und James Watson mit der Leitung betraut. Später wurde es in das „National Center for Human Research" umgewandelt. Dieses Zentrum war verantwortlich für die Koordination der anfallenden Arbeiten und die Verteilung der Gelder. Danach wurden auch Genomsequenzierungsprojekte in Japan und Europa begonnen und damit das International „Human Genome Sequencing Consortium" gegründet. Als größter Konkurrent stellte sich die Firma Celera mit ihrem Gründer Craig Venter dar. Der Wettlauf um das menschliche Genom hatte begonnen!
Zunächst mussten beide Gruppen menschliche DNA als Ausgangsmaterial zu Verfügung haben. Die Firma Celera nahm angeblich die DNA ihres Gründers Craig Venter. Das „International Human Genome Sequencing Consortium" (IHGSC) nahm die DNA eines Menschen, dessen Identität geheim gehalten wird.
Bei allen Sequenzierungsprojekten, also z. B. auch bei der Maus, wird die DNA zunächst in kleine Teile zerteilt und danach in sogenannte BACs (bacterial artificial chromosomes) kloniert. Im besten Fall ist nun pro BAC ein Teil der menschlichen DNA enthalten, während die Gesamtheit der BACs auch das gesamte menschliche Genom enthält. Diese BACs vermehren nun die menschliche DNA in Bakterien, z. B. im Darmbakterium E. coli. Die so vermehrte DNA wird dann sequenziert.
Was heißt nun eigentlich sequenzieren? Die Sequenz der DNA besteht im Grunde aus der Abfolge von vier Basen: Guanin, Cytosin, Thymin und Adenin. Man kürzt diese Basen mit ihren Anfangsbuchstaben ab, man schreibt also z. B. für Guanin ein G. Nach dem Sequenzieren erhält man also einfach eine Abfolge von Gs, Cs, Ts und As. Im ersten Schritt, der im Februar 2001 abgeschlossen war, erhielt man eine Rohsequenz. Dies war die Summe aller Teilsequenzen des menschlichen Genoms, die zu diesem Zeitpunkt bereits sequenziert worden waren. Man verwendete sogenannte Overlaps (= Überhänge) dazu, um die einzelnen Genstücke zusammenzufügen (Abbildung I). Lücken, sogenannte gaps, in den entstehenden Gesamtsequenzen werden durch das weitere Sequenzieren von humanen Genstücken in BACs (bacterial artificial chromosomes) aufgefüllt.
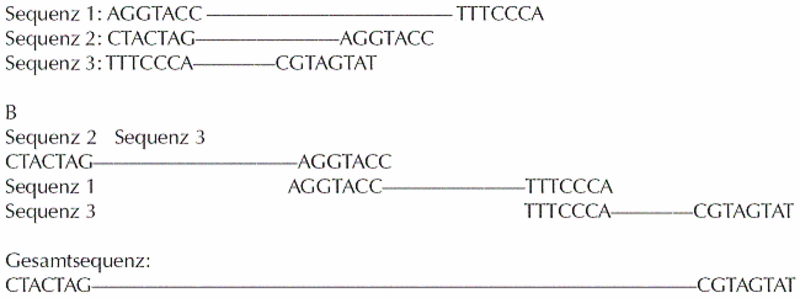
Allerdings ist es unmöglich, 100% des menschlichen Genoms auf diese Art zu sequenzieren, da einige Abschnitte auf der DNA auf Grund ihrer Struktur oder Sequenz nicht in Bakterien kloniert werden können, da sie zu fremdartig für das bakterielle System sind. Ein Ausweg aus diesem Dilemma ist die Verwendung von sogenannten YACs (yeast artificial chromosomes), da sie genauso wie der Mensch zu den Eukaryonten zählen und uns somit in der Verwendung von Strukturmotiven und Sequenzen in der DNA ähnlicher sind als die prokaryontischen Bakterien. Kouprina et al. konnten zeigen, dass die Stabilität von humaner DNA in YACS bei weitem höher war als in bakteriellen Systemen, außerdem wurden auch Stücke mit „problematischer" DNA kloniert.6
Dieses Füllen der Gaps war der zweite Schritt des Sequenzierungsprojekts, das im April 2003 sein Ende fand, mit der Verlautbarung, dass 98% nun durchsequenziert worden wären. Elbert Branscomb, einer der Direktoren des Joint Genome Instituts in Walnut Creek, das am Sequenzieren des menschlichen Genoms beteiligt war, meinte, dass dies das beste Ergebnis sei, das mit der heutigen Technologie erreichbar wäre. Einige Abschnitte sind schwer zu sequenzieren gewesen, da sie stark dupliziert waren und daher schwer in die menschliche Gesamtsequenz einzuordnen gewesen wären. Andere Teile waren aus unbekannten Ursachen nicht sequenzierbar. Vielleicht sind diese Abschnitte in einigen Jahren mit neuer Technologie aber durchaus zugänglicher.
Was wissen wir nun, da wir das menschliche Genom kennen?
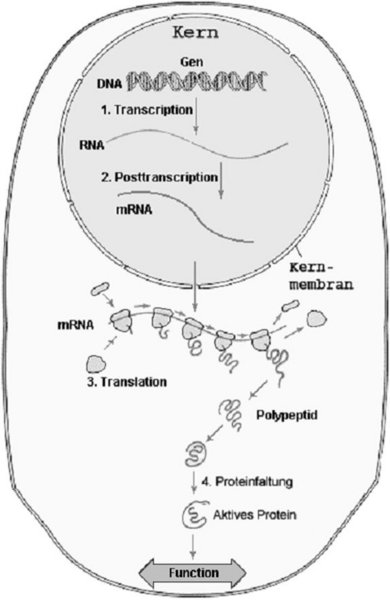
Wie schon bereits erwähnt ist die menschliche Sequenz nichts anderes als die Abfolge der vier Basen Adenin, Guanin, Cytosin und Thymin. Nach früheren Vorstellungen war diese Sequenz in Gene unterteilt. Über ein Zwischenprodukt, der Messenger- oder auf Deutsch Boten-RNA (mRNA), wurde aus dem DNA-Abschnitt ein Protein synthetisiert.
Dieses Modell, Gen – mRNA- Protein ist für viele Abschnitte der DNA gültig, d. h. viele Sequenzen codieren für Proteine. Man kann davon ausgehen, dass der Mensch ungefähr 30 000 Gene hat. Hier in diesem Modell hat sich auch der Begriff „genetischer Code" herausgebildet. Dieser Code ist im Grunde nichts anderes als die Umsetzung der DNA-Sequenz in Proteine. Wie vorhin schon kurz beschrieben, wird zunächst ein Zwischenprodukt namens mRNA synthetisiert, oder wie es im Fachjargon heißt „transkribiert". Diese mRNA erlaubt die Translation in ein Protein. Drei Basen der mRNA (ähnlich den Basen der DNA, nur dass statt Thymin die Base Uracil verwendet wird) codieren für eine Aminosäure. Was damit aber eigentlich gemeint ist, ist, dass ein Molekül namens tRNA diese drei Basen (Triplet) erkennt. An diesem kleinen Molekül befindet sich eine Aminosäure, die an die im Entstehen begriffene Aminosäurekette angehängt wird. Für jede tRNA Art, d. h. für jedes Triplet, gibt es eine Aminosäure.
Die Aminosäurenkette faltet sich dann in das Protein, das daraufhin seine Funktion aufnimmt (Abbildung II). Eine mögliche Funktion dieses Proteins ist die als Regulator in den vorher beschriebenen Prozessen Transkription und Translation. Man könnte nun vereinfachend sagen, dass ein gewisser Abschnitt auf der DNA für ein Protein codiert. So einfach ist es aber nicht. In den Abschnitten der DNA, die für Gene stehen, gibt es nämlich Teile, die aus der mRNA herausgeschnitten werden müssen, daher Bereiche, die nicht für die Proteinbildung nötig sind, bzw. sogar die Bildung eines funktionalen Proteins verhindern. Diese herauszuschneidenden Abschnitte heißen Introns, die proteincodierenden Teile sind die Exons. Was diese Introns sind, und warum sie in der DNA vorkommen, ist noch nicht vollständig geklärt. Einige Wissenschaftler vermuten, dass es sich bei den Introns um stillgelegte Transposons handelt. Einiges wie z. B. ähnliche Sequenzen mancher Introns zu Transposons scheinen diese Theorie zu bestätigen. Andere Introns hingegen übernehmen, nachdem sie aus der mRNA geschnitten wurden, regulatorische Aufgaben, meistens für das Gen, aus dem sie geschnitten worden sind. Im Übrigen ist ein großer Teil der DNA, die nicht für Gene codiert, dafür verantwortlich, RNAs zu bilden, die verschiedene wichtige Aufgaben übernehmen. Ich habe schon die tRNAs erwähnt, die für die Proteinsynthese (Translation) verwendet werden. Es gibt aber noch andere, oftmals sehr kleine RNA-Moleküle, die eine wichtige Rolle in der Regulation der Zellfunktionen spielen. Man nimmt auch an, dass die Unterschiede zwischen den Arten durch Unterschiede in diesen RNAs zustande kommen.7 Es wird noch viel Zeit vergehen, bis wir einiges über diese RNAs wissen. Eines ist aber auf jedem Fall klar: Wir haben es hier mit einem komplexen System zu tun.
Es ist also auf jeden Fall eine Vereinfachung, davon zu sprechen, dass die DNA ein Code ist, den es nur zu knacken gilt. Allerdings wurde, wie anfangs erwähnt, der Begriff genetischer Code als Modellvorstellung verwendet, um Unbekanntes durch Bekanntes zu erklären.8
Es ist also auf jeden Fall eine Vereinfachung, davon zu sprechen, dass die DNA ein Code ist, den es nur zu knacken gilt. Allerdings wurde, wie anfangs erwähnt, der Begriff genetischer Code als Modellvorstellung verwendet, um Unbekanntes durch Bekanntes zu erklären.8
Aussichten
Aber auch für die 30.000 Sequenzabschnitte auf dem menschlichen Genom, die für Gene codieren, kann nicht gesagt werden, dass wir viel darüber wissen. Wir haben es hier ebenfalls mit einer Ansammlung von Daten zu tun, die erst durch weitere Forschung in Wissen umgewandelt werden kann.9 Es ist sehr wichtig, zwischen Daten und Wissen, die ein Experiment liefert, zu unterscheiden. Viele dieser Gene sind noch unbekannt, d. h. die Funktion, die sie ausführen, ist noch nicht erkannt worden. Ein Forschungsfeld, das sich „functional genomics" nennt, versucht nun die Funktionen neuer Gene herauszufinden. Für einige dieser als Gene identifizierten Abschnitte gibt es homologe Gene in der Maus oder in anderen Modellorganismen (Huhn, Fruchtfliege, Zebrafisch…), und man kennt daher von dort ihre Funktion. Allerdings gibt es auch Fälle, dass ein Gen in dem einen Modellorganismus eine wichtige Funktion erfüllt und in einem anderen nicht. Es ist also auch mit diesem Wissen noch nicht gesagt, ob beim Menschen das homologe Gen zur Maus dieselbe Rolle spielt. Man hat z. B. in der Maus festgestellt, dass Weibchen mit einer gewissen Ausprägung eines Gens mit sehr hoher Wahrscheinlichkeit an Brustkrebs erkranken. Man nahm nun an, dass es bei Frauen einen ähnlichen Zusammenhang geben könnte. Es ist zwar tatsächlich so, dass die Frauen mit diesem bestimmten Gen eine erhöhte Wahrscheinlichkeit haben, an Brustkrebs zu erkranken, aber diese Wahrscheinlichkeit liegt bedeutend unter jener der Mäuse. Man weiß noch nicht, welchen Einfluss die Umwelt (Nahrung, soziales Umfeld etc.) spielt. Das Feld der Epigenetik untersucht diese Wechselwirkung zwischen Genen und der Umwelt.
Allerdings ist es problematisch, wenn in der Öffentlichkeit bekannt wird, dass das Gen für Brustkrebs oder auch für Alzheimer oder sonst eine genetisch bedingte Krankheit gefunden wurde. Versicherungen, so die Befürchtung vieler, wären natürlich interessiert, solche Leute mit dem Risiko einer Krankheit, nur über höhere Beiträge zu versichern. Dies wäre eine neue Form der Diskriminierung. Hinzu kommt, dass noch nicht klar feststeht, ob bei diesem Menschen die Krankheit auch wirklich ausbricht.
Eine weitere Befürchtung ist, dass die Schere zwischen Diagnose und Therapie noch weiter auseinander klafft. Wie soll man vorgehen, wenn bei einem Embryo ein Erbdefekt festgestellt wird, der noch nicht heilbar ist? Wie soll man einem Patienten klarmachen, dass er mit einer gewissen Wahrscheinlichkeit an einer Form von unbehandelbarem Krebs erkranken wird, es aber durchaus möglich sein kann, dass dieser Krebs nie ausbricht?
Eines ist aber auf jeden Fall sicher: Obwohl wir nun die Sequenz des menschlichen Genoms kennen, wird noch einige Zeit vergehen, bis uns tatsächlich klar ist, was für Wissen und therapeutischen Nutzen wir aus ihr und der weiteren Forschung erwerben können.10 Es ist aber auf jedem Fall gut, sich schon jetzt über die Verwertung dieses Wissens Gedanken zu machen.
Glossar
BAC (bacterial artificial chromosome): Künstliches bakterielles Chromosom. Erlaubt die Vermehrung jeder gewünschten DNA-Sequenz in Bakterien. Dies ist günstig und schnell möglich, da sich Bakterien schnell vermehren.
Eukaryonten: Besitzen einen echten Zellkern, der die DNA umschließt.
Genom: Die Summe aller Gene eines Lebewesens.
Prokaryonten: Besitzen keinen Zellkern. Die DNA liegt quasi frei in der Zelle.
Transposon: Mobile DNA Stücke. Sie können sich vermehren (replizieren) und auch ins Genom eines Wirtes integrieren. Später können sie sich wieder von dieser Stelle herausschneiden und woanders integrieren. Manchmal passiert es dabei, dass sie Teile des Wirtsgenoms mitnehmen und so zu Mutationen führen.
Referenzen
- Pearson H., Human genome done and dusted, Nature Science Update (April 2003)
- Schwarz M., Humanes Genom Projekt, Imabe-Dokumentation 1/1991
- Cokk-Deegan R. M., The Alta summit, Genomics (1984); Vol 5: 661-663
- Sinsheimer R. L., The Santa Cruz Workshop, Genomics (1985); Vol 5, 954-956
- Dulbecco R., A turning point in Cancer Research: Sequencing the Human Genome, Science (1990); Vol 231: 1055-1056
- Kouprina N. et al., Segments missing from the draft human genome sequence can be isolated by the transformation-associated recombination cloning in yeast, EMBO Reports (2003); Vol 4: No 3
- Mattick et al., The Evolution of Controlled Multitasked Gene Networks: The role of introns and other noncoding RNAs in the Development of complex Organisms, Mol Biol Evol (2001); Vol 18: 1611-1630
Mattik J. S., Non-coding RNAs: the architect of eukaryotic complexity, EMBO Reports (2001); Vol 2: No 11 - Kay L. E., Who wrote the book of life? The history of the genetic code, Stanford (2000)
- Duyk G. M., Sharper tools and simpler methods, Nature Genetics (2003); Vol. 32: 465-468
- Honnefelder L., Was wissen wir, wenn wir das menschliche Genom kennen? in: Die Herausforderung der Humangenomforschung. Eine Einführung, DuMont Verlag, Köln (2001), S. 9-25
Mag. rer. nat. Carmen Czepe
Institut für Genetik
Universität Köln
Weyertal 121, D-50931 Köln



